
Корнелюк Олексій Сергійович
Лікар загальної практики-сімейний лікар
«Медичний центр «Салютем»
Україна, м. Вінниця
Маліновська Наталія Михайлівна
Студентка кафедри медицини та реабілітації
Вінницький соціально-економічний інститут Вищого Навчального Закладу
Відкритий міжнародний університет розвитку людини «Україна»
Україна, м. Вінниця
Анотація. Метою даної статті є висвітлення патофізіологічних процесів ВІЛ-інфекції: вивчення основ будови вірусу імунодефіциту людини; особливості проникнення ВІЛ у людський організм; перебіг фаз ВІЛ-інфекції та перехід її у СНІД.
Ключові слова: ВІЛ, ВІЛ-інфекція, HIV, СНІД, AIDS, патофізіологія.
Вірус імунодефіциту людини спричиняє імунодефіцит, що характеризується зниженням кількості Т-хелперів (CD4+ клітин). Втрата CD4+ клітин призводить до розвитку опортуністичних інфекцій та неопластичних процесів.
ВІРУСОЛОГІЯ ВІЛ
ВІЛ-1 та ВІЛ-2 є ретровірусами з родини Ретровіруси, роду Лентівірус. Містять диплоїдний геном - дві копії одноланцюгової, позитивно-смислової (+) РНК. ВІЛ перетворює свою РНК у ДНК-копію (яка є по суті провірусом) та вбудовує її у клітину-хазяїна.
ВІЛ містить 3 структурних, «групоспецифічних» гени: gag, pol та env. Ген gag (від group-specific antigens) кодує внутрішні, структурні протеїни, зокрема, поліпротеїн p55, який вірусна протеаза розщеплює на структурні білки p6, p7, p17 та p24. Ген pol (від polymerase – полімераза) кодує протеазу (PR), інтегразу (IN) та зворотну транскриптазу (RT). Ген env (від envelope glycoprotein – глікопротеїн оболонки) кодує оболонку вірусу, протеїн gp160, який розщеплюється фурином на структурні типоспецифічні білки gp41 та gp120. Глікопротеїн 120 (gp120) і є тим вірусним білком, що взаємодіє з клітинним рецептором CD4 клітиини (малюнки 1-4).
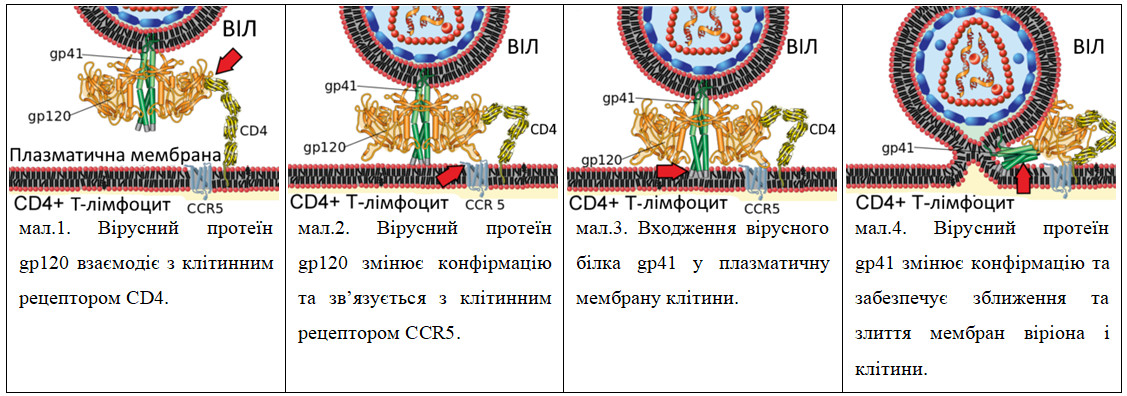
Також ВІЛ містить три регуляторні гени, що забезпечують контроль за реплікацією вірусу. Ген tat (transactivator of transcription – трансактиватор транскрипції) пов’язує вторинну структуру геномної РНК так активує її зворотну транскрипцію, активує синтез вірусних матричних РНК та регулює вихід віріонів із заражених клітин. Ген rev (regulator of virus expression – регулятор експресії вірусних генів) регулює експресію білків віріона, забезпечує транспортування елементів вірусу з ядра і відповідає за зміну синтезу регуляторних білків вірусу на синтез структурних. Обидва ці гени необхідні для трансляції, оскільки вони забезпечують стимуляцію транскрипції провірусної ДНК та переніс РНК у цитоплазму. Ген nef (negative expression factor – негативний фактор експресії) дозволяє вірусу уникати атаки Т-кілерів і розпізнавання клітинами CD4+. Також цей ген пригнічує активацію Т-лімфоцитів шляхом зв’язування різних білок-компонентних систем сигнальної трансдукції.
Ще одна група генів ВІЛ – додаткові гени. Ген vif забезпечує повноцінну і правильну реплікацію вірусу, оскільки попереджають мутаційні заміни у клітинах-мішенях. Штами без цього гену здатні до проникнення у клітини-мішені і початок зворотної транскрипції, але синтезована такими штамами вірусна ДНК не є завершеною. Ген vpr є специфічним для лентівірусів і забезпечує реплікацію вірусу у неподільних клітинах, наприклад, макрофагах. Ген vpu є специфічним для ВІЛ-1 [3] (у ВІЛ-2 – vpx) і забезпечує руйнування клітинного рецептора CD4 та стимуляцію виділення дочірніх віріонів за рахунок пригнічення або ж повної деактивації інтерферон-індукованого трансмембранного білка тетерину.
БІОЛОГІЧНІ ОСНОВИ СНІДу
Механізм розвитку хвороботворного процесу, що призводить до СНІДу, до цих пір не є до кінця вивченим [4].
Спостерігається специфічне зниження рівня CD4+ клітин, що призводить до дисбалансу CD4/CD8 клітин та порушенню регуляції синтезу антитіл B-клітинами. Імунна відповідь до деяких антигенів знижується, і інфікована людина стає неспроможною адекватно реагувати на патогенні та умовно-патогенні мікроорганізми. Оскільки порушеною є здебільшого клітинна ланка імунітету, переважають інфекції небактеріальної природи (вірусні або грибкові).
Картина опортуністичних інфекцій у розрізі регіонів відображає найбільш поширені інфекції даному регіоні. Так, у країнах, що розвиваються, найбільш поширеною опортуністичною інфекцією є туберкульоз, у США – пневмоцистна інфекція та грибкові ураження. В той же час, наприклад, серед чоловіків-гомосексуалістів поширена саркома Капоші, що обумовлено супутнім інфікуванням вірусом герпесу людини 8 типу.
Також у реплікації вірусу відіграє роль лімфоїдна тканина травної трубки [5]. Хоча пусковим механізмом для ВІЛ-інфекції зазвичай слугує прямий контакт із кров’ю або експозиція вірусу на слизових оболонках геніталій, ШКТ є ідеальною структурою для реплікації вірусу через велику кількість лімфоїдної тканини. І, оскільки контролювати вірусну реплікацію в ШКТ дуже важко, часто антиретровірусна терапія не може забезпечити тривалий контроль над інфекцією [6, 7]. Вірусна реплікація проходить у всіх відділах ШКТ. Згідно даних, отриманих дослідниками у 2003 році [8], існує припущення, що раннє лікування здатне призвести до кращого відновлення CD4+ клітин у лімфоїдній тканині ШКТ, проте інші клінічні дані поки що не підтверджують ці судження.
На додачу, активну реплікацію ВІЛ іноді виявляють і у пацієнтів з нібито пригніченою реплікацією, про яку судять по вірусному навантаженню у плазмі крові. Т-кілери (CD8+ клітини) реагують на ВІЛ у лімфоїдній тканині травної трубки і ця реакція під час антиретровірусної терапії не знижується так само, як під час «периферійних» вимірювань. Ці дані підкреслюють обмежене використання «периферійних» вимірювань, і показують що ж насправді являє собою «центральна» реплікація ВІЛ.
Однією з теорій, яка пояснює таку різницю у вимірюваннях у травній трубці та периферійній крові, є те, що реплікація вірусу та імунна активація можуть фактично ускладнювати ефективне поповнення CD4+ клітин. Дослідження реплікації Т-клітин показали, що при нелікованій ВІЛ-інфекції наявна швидка циркуляція дефектних Т-клітин (з тимуса) [9]. До цих пір немає єдиної думки, спричинені ці дефекти та швидкість циркуляції безпосередньо прямою дією вірусу, або ж особливістю імунної відповіді на ВІЛ-інфекцію.
Відомо, що для нормального співвідношення цитокінів необхідний нормальний клітинний цикл, а ВІЛ спричиняє зупинку останнього. Аналіз цитокінового профілю у ВІЛ-інфікованих, здорових та лікованих пацієнтів виявив чіткі зміни у рівнях клітин імунітету. Зокрема, у інфікованих спостерігалось зниження рівнів ІЛ-7, ІЛ-12, ІЛ-15 та ФРФ-2, а також підвищення рівнів ФНП-а та ІФНу-індукований білок 10 (IP-10). Також аналіз показав, що антиретровірусна терапія частково виправляла ці відхилення [10].
Білки (гени) ВІЛ безпосередньо впливають на функціонування Т-клітин, порушуючи клітинний цикл. Тож, втрата Т-клітин є першочерговою проблемою, оскільки вона призводить до зменшення кількості антигенів, на які здатна реагувати імунна система інфікованого. Антиретровірусна терапія здатна зупиняти цей процес, але ступінь зворотності є вищим, якщо терапію розпочинати якомога раніше; і стрімко знижується, якщо терапію розпочинають при рівні CD4 Т-клітин 200/мкл і нижче.
Пряма цитотоксична дія вірусної реплікації не є першопричиною втрати CD4 Т-клітин. Такий ефект є, скоріше за все, відображенням Т-клітинного апоптозу як частини імунної гіперактивації у відповідь на хронічні інфекції. Інфіковані клітини також можуть бути уражені імунною відповіддю.
Тож, незалежно від власне причини порушення, втрата механізму тимічного заміщення в умовах індукованої імунної активації разом із втратою Т-клітин і є ключовим компонентом зменшення репертуару Т-клітин і прогресуванню ВІЛ-інфекції у СНІД.
ВІЛ-інфекція спричиняє видимі порушення у структурі лімфовузлів. Такі порушення враховуються при визначенні стадії захворювання [11].
ФАЗИ ВІЛ-ІНФЕКЦІЇ
Клінічно можна виділити 3 основні стадії: гостра сероконверсія, безсимптомна інфекція та СНІД. Деякі науковці виділяють окремо стадії інфікування та ранніх проявів ВІЛ-інфекції.
Фаза гострої сероконверсії. Під час моделювання на тваринах було відмічено, що клітини Лангерганса є першими клітинами-мішенями для ВІЛ. У людей виникнення віремії з широким розповсюдженням вірусу зазвичай спостерігається через 4-11 днів після проникнення вірусу через слизові оболонки.
Зазвичай немає певного місця прикріплення вірусу. ВІЛ прикріплюється там, де є багато вільного хроматину та більш доступна ДНК. На цьому етапі формується резервуар провірусної ДНК. Вимірювання рівнів копій ДНК показали, що такі рівні є досить стабільні.
ВІЛ на своєму шляху зустрічає різні біологічні рідини, як мають відповідно різні ступені агресивності. Так, слина та шлунковий сік руйнують віріони ВІЛ більше за інші біологічні рідини.
Розмір провірусного резервуару є прямопропорційним вірусному навантаженню та обернено-пропорційним відповіді CD8+ Т-клітин. На такому етапі агресивне раннє лікування дає великі переваги у довготривалій перспективі.
Далі, разом із зростанням вірусного навантаження, швидко зменшується кількість CD4+ Т-клітин. З появою антитіл до ВІЛ та реакції CD8+ Т-клітин, вірусне навантаження спадає до певного стійкого рівня, а кількість CD4+ Т-клітин повертається до попередніх рівнів, іноді навіть у межах норми.
Сероконверсія може займати від кількох тижнів до кількох місяців. Проміжок часу від зараження до появи антитіл називається «сліпим вікном» (серонегативним вікном). Симптоми під час сероконверсії можуть включати гарячку, грипоподібні стани, лімфаденопатії та висипки. Зустрічаються приблизно у половини ВІЛ-інфікованих.
Латентна фаза, або фаза безсимптомної інфекції. Під час цієї фази у людини може не бути жодних клінічних ознак або симптомів дуже тривалий час, інколи більше десяти років. Протягом цього періоду відбувається реплікація вірусу, імунна відповідь організму на нього зберігається, але поступово знижується. За відсутності лікування, вірусне навантаження має тенденцію до стабільності, тоді як рівень CD4+ Т-клітин зменшується.
Щодо безпосереднього механізму реплікації вірусу у клітинах організму-хазяїна слід виділити такі моменти:
- Проникнення у клітину шляхом взаємодії gp120 та корецептора CXCR4 або ж CCR5 [12]. На цьому етапі можуть бути ефективними препарати, що блокують корецептори.
- Взаємодія gp41 з мембраною, що призводить до злиття віріона та клітини-мішені.
- Проникнення вмісту віріона у клітину.
- Вивільнення РНК з капсиду.
- Зворотна транскрипція під дією зворотної транскриптази. На цей етап діє переважна більшість препаратів, затверджених для лікування ВІЛ-інфекції.
- Активація CD4+ лімфоцитів при контакті їх з антигенпредставленими клітинами.
- Інтеграція провірусної ДНК у активні лімфоцити. На цьому етапі застосовують препарати, що інгібують інтегразу.
- Синтез попередників матричних РНК в ядрі. Його трансформації.
- Синтез вірусних білків (трансляція) в цитоплазмі клітини.
- Збір геномних РНК вірусу та вірусних білків-попередників вздовж мембрани клітини.
- Розщеплення протеазами вірусних білків-попередників до функціональних компонентів з формуванням зрілих віріонів. На цей етап діють препарати, що інгібують протеази.
- Зрілі віріони відділяються від клітини-хазяїна з частиною її мембрани і виходять в кров’яне русло, натомість клітина хазяїна гине.
Далі віріон перебуває у судинному руслі до 8 годин, що дозволяє йому активно шукати нові місця для прикріплення та реплікації.
СНІД. Коли враження імунної системи досягає критичного рівня і починають розвиватися опортуністичні інфекції, ВІЛ-інфекція переходить у синдром набутого імунодефіциту. У більшості країн критерієм діагностики СНІДу є рівень CD4+ Т-клітин ≤200/мкл. Проте описано розвиток опортуністичних інфекцій при рівні CD4+ клітин більше такого показника, в свою чергу, деякі люди з рівнем CD4+ Т-клітин менше 200/мкл не інфікуються іншими захворюваннями.
Загалом, багато опортуністичних інфекцій та станів враховуються при переході ВІЛ-інфекції у СНІД. Частота таких інфекцій у популяції варіюється від рідкісних до дуже поширених. Проте, у імунокомпетентних осіб, всі опортуністичні інфекції зустрічаються нечасто або проходять у легкій формі. Коли ж одна з таких інфекцій переходить у важку форму або стає дуже частою, і немає інших пояснень імунодефіциту, мова йде про діагноз СНІД.
ІМУНОЛОГІЧНИЙ КОНТРОЛЬ ВІЛ-ІНФЕКЦІЇ
Основним механізмом імунологічного контролю ВІЛ є цитотоксичні CD8+ Т-клітини. Т-клітинні відповіді корелюють із станом вірусного навантаження та швидкістю прогресування. Клітинний імунітет є визначальним фактором у людей, які неодноразово контактували з ВІЛ, але не інфікувалися.
Хоча у інфікованих і виявляють антитіла проти ВІЛ, очевидно, що їх дії недостатньо, щоб допомогти контролювати інфекцію.
Для початкового контролю ВІЛ досліджується роль NK-клітин (натуральних кілерів).
Література:
1. Shelley A Gilroy, John J Faragon, HIV Infection and AIDS. Medscape. 2020 Mar 05. 5:9-48
2. Наказ МОЗ України від 05.06.2019 № 1292 "Про затвердження нового Клінічного протоколу із застосування антиретровірусних препаратів для лікування та профілактики ВІЛ-інфекції"
3. Gao F; Bailes E; Robertson DL, et al. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature. 1999 Feb 4. 397(6718):436-41
4. Weber J. The pathogenesis of HIV-1 infection. Br Med Bull. 2001. 58:61-72.
5. Talal AH; Irwin CE; Dieterich DT; Yee H; Zhang L. Effect of HIV-1 infection on lymphocyte proliferation in gut-associated lymphoid tissue. J Acquir Immune Defic Syndr. 2001; 26(3):208-17
6. Poles MA; Boscardin WJ; Elliott J, et al. Lack of decay of HIV-1 in gut-associated lymphoid tissue reservoirs in maximally suppressed individuals. J Acquir Immune Defic Syndr. 2006; 43(1):65-8
7. van Marle G; Gill MJ; Kolodka D; McManus L; Grant T; Church DL. Compartmentalization of the gut viral reservoir in HIV-1 infected patients. Retrovirology. 2007; 4:87
8. Guadalupe M; Reay E; Sankaran S; et al. Severe CD4+ T-cell depletion in gut lymphoid tissue during primary human immunodeficiency virus type 1 infection and substantial delay in restoration following highly active antiretroviral therapy. J Virol. 2003; 77(21):11708-17
9. Bandera A; Ferrario G; et al. CD4+ T cell depletion, immune activation and increased production of regulatory T cells in the thymus of HIV-infected individuals. PLoS One. 2010; 5(5):e10788
10. Keating SM; Golub ET; Nowicki M; Young M et al. The effect of HIV infection and HAART on inflammatory biomarkers in a population-based cohort of women. AIDS. 2011; 25(15):1823-32
11. Edén A, et al. HIV-1 viral escape. J Infect Dis. 2010 Dec 15. 202(12):1819-25.
12. Eoin Coakley, et al. Assessing Chemokine Co-Receptor Usage in HIV. 2005 Feb;18(1):9-15